FAQs
Equipment (Moberg, PbtO2, etc.)/Trial Materials
ICP Placement Procedure and Pbt02 Probes
Subject Reimbursement/Finances
Q: What do you do if a participant says they want to withdraw from the study?
A: The goals in responding to this kind of inquiry are to make sure participants know and understand their options, that we respect the choice of the participant, and that we preserve the integrity of data collection as much as possible.
- Do not pressure the participant (or LAR) to remain in the study. A research participant always has the right to withdraw from the study at any time and for any reason.
- Often your site PI will be in the best position to discuss options, as well as the importance of participation. Ask if the participant (or LAR) would like to talk with the PI.
- Inform the PI as soon as is practical either way.
- Try to clarify what they want to stop. Often requests to withdraw from the study are just requests to withdraw from certain elements of the intervention, or to skip a blood draw or other assessment or visit. Participants may opt out of most elements without withdrawing from the study.
- Ask specifically if they would like to completely withdraw from the study or are reticent about a particular part of the study (the outcome evaluation, monthly phone calls, for example).
- Study personnel should be aware that we can usually forgo the more burdensome portions of the study.
- If they are amenable to a short phone call in 6 months to see how they are doing, let them know that you can speak with the LAR, a friend or relative, or them. The study’s primary goal is to obtain the GOSE at the 6-month post-injury point.
- Ask “even If you want to stop everything else, may we look at your medical record while you are in the hospital?” Even if they only agree to continued chart review, this should not be considered a withdrawal of consent for participation.
- If they want no further contact, honor their wishes and thank them. Document withdrawal of consent on the end-of-study case report form.
As you talk with the participant/family it may become clear that they do not want to participate under any circumstance. In this case it is certainly their right to withdraw and no further contact should be made by study staff. Thank them for their participation.
Q: What if a subject tests positive for COVID?
A: Patients do not need to be screened for COVID-19 infection in order to participate in BOOST3. Enrollment of eligible patients in BOOST3 should never be delayed because of institutional COVID-19 screening requirements. Asymptomatic or minimally symptomatic COVID-19 infection, whether diagnosed before or after enrollment does not alter eligibility for, or continued participation in, BOOST-3. As in other forms of pneumonia, impairments in oxygenation prior to enrollment may affect eligibility of those with more severe infection. Pre-existing COVID-19 infection diagnosed after enrollment is not a reportable AE in BOOST unless severity markedly worsens. COVID-19 infection thought to have been acquired after enrollment may be reportable as an adverse event if it causes an illness that requires treatment.
Q: What are the documentation requirements for capture of Tier therapies during the monitoring period?
A: It is the study personnel’s responsibility to examine all options used for documentation to ensure that any intervention used to address low brain oxygen or raised ICP was performed and documented into the Moberg CNS monitor. As a study coordinator, if it is apparent that an intervention was performed but not documented in the Moberg CNS system, it will need to be added at a later time. Study coordinators should assess for this during their daily check on a BOOST3 patient during the monitoring period. Upon initial use, it will be important for the study coordinators to be present at the bedside to make sure that the nurses are comfortable with the options presented for data capture for BOOST3. Three documentation options are listed below:
Medical Record: From a patient care/clinical perspective, clinical nurses should be documenting (any) interventions done for patient care in the medical record; for example, a change in PEEP should be documented along with the rationale.
Moberg CNS Monitor: The Moberg CNS monitor will be used for BOOST 3 subjects for data capture. An alarm will be programmed into the monitor to alert the nurse when a sustained (> 5 minutes) ICP or PbtO2 abnormality is detected; at minimum, the nurse should acknowledge the alarm using the touch screen function on the CNS monitor. Once the alert is acknowledged, the Moberg monitor will display a menu of Tier 1 interventions to address the abnormality. This should trigger the need to perform a Tier 1 intervention—the nurse will need to select the intervention chosen to address the abnormality and perform the intervention, and then document the time that the intervention was performed using the touch screen function on the Moberg, as these interventions will be programmed into the CarePath software.
Nursing Documentation Tool (OPTIONAL): An optional paper Nursing Documentation Tool was developed for nurses who prefer to document Tier Interventions on paper vs interacting with the CNS monitor software (they will still have to acknowledge/silence the alarm), OR if there is a technical issue with the CNS monitor. This tool lists all of the tier interventions for each Scenario. In the event of an abnormality, depending on the scenario, the nurse can simply write their initials in the appropriate column that corresponds with the tier intervention performed, and document the date and time using the far left column. The study team should provide a new set of papers either each shift, or day the subject is being monitored. If a nurse chooses to document using the Moberg, they are NOT required to double document on the Nursing Documentation Tool.
NOTE: The Moberg has a feature in which the nurse can review events/interventions that were documented throughout their shift. If the nurse had a busy shift, they can simply use the review events feature to quickly chart in the medical record the interventions that were performed for their patient that will also include the correct time the intervention was performed. They can also do this with the Nursing Documentation tool, if used in place of Moberg, especially if the paper tool is available in the room so that they can quickly chart interventions.
Q: When does a scenario begin?
A: A Scenario begins at the time an abnormality is noted on the Moberg CNS monitor or on the ICP and/or PbtO2 monitor. The Moberg alarm will sound when there is an abnormality. It is generally accepted that an abnormality sustained for 5 minutes is a true abnormality that requires a response by the medical care provider. However, it is not necessary to wait 5 minutes from the time the abnormality began in order to respond if the provider believes the abnormality is valid. The medical care provider may use his or her best judgment as to whether or not to wait 5 minutes from the beginning of the abnormality to respond. The protocol requires treatment initiation within 15 minutes from the very beginning of the abnormality. The Scenario begins at the time the value became abnormal (not after 5 or 15 minutes). A Scenario ends after the value is normal for 30 minutes from the time it became normal. It needs to be normal for 30 minutes to end that Scenario and begin a new one when the next abnormality occurs.
Q: How do we record 2 abnormalities that occur within 30 minutes?
A: If an abnormal ICP or PbtO2 is corrected and then becomes abnormal again within 30 minutes, treat as one Scenario. May select treatments from Tier 1, 2, 3.
Example: ICP was reduced after initiating treatment from Tier 1 in 10min, but back up within 30 minutes. These two Scenarios (2 abnormalities) occurred within 30 minutes and should be considered one Scenario.
Q: What is required for a NEW Scenario to begin?
A: If you have already been working in a Scenario (B, C, or D) to correct an abnormality (or abnormalities) and the treatments successfully corrected the abnormalities for minimum of 30 minutes that Scenario is ended. Remember that the abnormal ICP and PbtO2 that began the Scenario must be within NORMAL limits for AT LEAST 30 MINUTES. Then if another abnormality occurs, begin a NEW SCENARIO. You must move to a tier 2 treatment at 1-hour if the value has not normalized at all during that hour.
Q: What are common mistakes regarding the FiO2 challenge?
A: Not doing one or doing it too late (> 2 hours after placement).
Remember to record the following:
- The FiO2 before beginning the challenge
- The time at the beginning of the challenge
- The FiO2 number at the end of the challenge
- The time at the end of the challenge
- Whether it was successful (rose at least 5 mm Hg within 20 min. maximum).
ICP Placement Procedure and Pbt02 Probes
Q: How long will placement of the ICP and PbtO2 probes take? Where will this procedure be performed?
A: The procedure takes approximately 20-30 minutes. This procedure is performed most commonly in the intensive care unit. In some occasions it is performed in the operating room if the patient requires a surgical procedure. In rare cases, in some institutions, the probes are placed in the emergency department.
Q: Will this be an inpatient or outpatient procedure?
A: This is an inpatient treatment protocol with all procedures being done in the hospital setting.
Q: Will there be anesthesia required for placement of intracranial monitors?
A: Yes. In the intensive care unit or emergency department, the probes are placed under local anesthesia with moderate or unconscious sedation protocols. If the probes are being placed in the operating room, there is no need for additional sedation or anesthesia as they are typically being placed under for other surgical procedures that are determined to be necessary by the treating neurosurgeon.
Q: Why are children included in BOOST-3?
A: Children aged 14 years and older will be enrolled in the study. We plan to exclude children younger than age 14 years for several reasons. First, the outcome measures proposed are not fully validated in young children. Second, pre-pubertal children have a distinct cerebrovascular physiology, and in particular are at greater risk of hyperemia and malignant intracranial hypertension. This may place them at greater risk of PbtO2 directed therapies and may confound the interpretation of our results.
Q: Will BOOST-3 allow for co-enrollment into any other studies?
A: Clinical studies may be considered for co-enrollment into BOOST-3 as long as there is approval from the study leadership for both studies (examples include EPI-BIOS, Spreading Depolarizations II, PPOWER, and SDII) ahead of time. Sites should consider any potential logistical concerns before a decision is made to allow for co-enrollment and should be reviewed with local Site PIs. The decision to allow co-enrollment depends on the ability to preserve the scientific integrity of both studies.
NOTE: There is NO co-enrollment of patients in TXA.
Protocol/Protocol Adherence (New 6-2-21)
Q: Is it required that the treatment team always adhere to the protocol?
A: Yes, adherence to the protocol is required. There is substantial room within each Scenario and within each Tier for neurointensivists, neurosurgeons, and other treatment team members at each site to choose the particular intervention to implement, which should be informed by the detailed knowledge of the pathophysiology active in a particular patient at a particular time. The treatment team may choose not to do interventions responding to an abnormal PbtO2 or ICP for medical/safety reasons. However, for Tiers 1 and 2, the available treatment options are broad enough that it would be highly unusual that all of the treatment options would be contraindicated in a given patient. Tier 3 treatments are optional in all cases and are included in the CRFs so we can record therapies that could be instituted. If site investigators chose not to implement any Tier 1 or Tier 2 therapies, the rationale for that decision needs to be clearly documented in the medical chart and recorded by the research team to avoid protocol violations.
Q: Is it ok to disable the alarm on the ICP/PbtO2 monitors?
A: Only the PbtO2 alarm in the blinded group should be disabled. Do not disable the alarms in the unblinded (PbtO2 and ICP treatment) group. If the alarms are disabled, abnormalities will be missed in the open treatment group.
Q: Is it ok to alter the ICP and/or PbtO2 thresholds on the external monitors in order to minimize the number of times the alarms are sounding?
A: No. Coordinators need to check the monitors every morning in order to ensure that the treatment staff is not altering the threshold numbers of the ICP/PbtO2 monitors. The PbtO2 monitor alarm MUST be silenced prior to the cover being placed if the participant is in the blinded group so that the treatment team will not be made aware of PbtO2 abnormalities.
Q: What is the length of time in which subjects must be randomized into BOOST-3?
A: Eligible subjects must be randomized within 6 hours of arrival to the enrolling hospital, and no later than 12 hours from injury to qualify for study enrollment. In order to be randomized, the ICP and PbtO2 monitors must be placed.
Q: What about patients who require immediate surgical evacuation of a hematoma? Do monitors still have to be inserted within 6 hours?
A: Yes. If it is not feasible to place monitors and randomize within 6 hours of arrival to enrolling hospital (and within 12 hours from injury), the patient will be ineligible for study enrollment. In cases where subjects are taken to the OR for a surgical intervention, monitors can be placed in the OR and randomization should take place during this time, provided it’s within 6 hours of arrival. Treatment can start when it is practical and feasible to do so.
Q: If a patient is in BOOST-3, are we allowed to use the Hemedex at all?
A: No. Neuro-monitoring devices that are not acceptable include: NIRS, Jugular bulb saturation monitors, cerebral oximetry monitors, cerebral blood flow monitors, continuous transcranial doppler (TCD) monitoring, or quantitative EEG algorithms specific for cerebral hypoxia, as these can provide indirect information regarding brain tissue oxygenation. The occasional use of TCDs to confirm suspected vasospasm or assess autoregulation is allowed. The use of quantitative EEG for detection of seizures is acceptable. If vasospasm or seizures are identified by any means, this information must be documented in the Adverse Event CRF.
Q: When is the best time to randomize a patient?
A: The best time to randomize is as soon as possible, after the monitors are placed. Keep in mind that for a subject to be eligible for enrollment, monitor placement must occur within 6 hours of hospital arrival, and no later than 12 hours following time of injury. If there is a situation in which a patient who you know will be getting a monitor but there is a potential delay (such as a surgery), the patient should not be randomized until you can confirm that the monitor was placed within the protocol defined window. If the monitors are placed outside of the protocol defined window, then the subject will no longer be eligible for the study. A subject is considered ‘enrolled’ when the randomized treatment allocation is generated. If an LAR signs a consent but the subject is not able to be randomized in time or does not receive monitors in time, they will be captured as a ‘screen failure’ subject.
Q: What are the guidelines for establishing time of injury?
A:
- Look for documentation if event was witnessed or unwitnessed
- If unwitnessed, look for evidence of when the patient was last observed as well
- If witnessed event, defer to witness recollection of time of event, or estimate based on when EMS is called
- If time of injury is estimated and not documented in the medical record, write a note to file to use as your source document for monitor review.

Q: Is the SIREN e.consent platform part 11 compliant?
A: Yes. SIREN uses the University of Michigan implementation of the REDCap platform to obtain electronic consent and (in some SIREN studies) participant contact information in a process that is 21 CFR Part 11 compliant. Part 11 compliance requires information technology that is capable of supporting compliant processes, and specific operating procedures that use the IT appropriately. Documentation from the University of Michigan Health Information Technology and Services (HITS) of the physical and network information technology security that supports Part 11 compliance in this implementation is available here. Documentation that the SIREN processes are Part 11 compliant is found in the SIREN Network e.consent SOP.
Q: Who are the personnel at the SIREN CCC and who should we communicate with for various trial related questions?
A: The easiest way to reach out to the SIREN CCC is to use the BOOST-contact@umich.edu. This will ensure that your message gets to the appropriate team member who can provide a prompt response.
Q: How can we confirm prospective trial sites, affiliated SIREN Hubs and critical study team member contact information with the SIREN CCC?
A: This is a great question, as we want to be sure that we have the contact information to reach all critical study team members at all prospective BOOST 3 sites. If there have been any changes to your site's Hub affiliation, BOOST 3 PI, or BOOST 3 Primary Study Coordinator since you completed the initial survey, please let us know. We are happy to work with you to confirm this information.
Q: What if a treatment that is listed in the Tiers is already being done prior to the beginning of an abnormality that signals the beginning of a Scenario? (An episode is sometimes referred to as a Scenario. It is the period of beginning of an abnormality and the treatment period). Is there an exception in which you may begin in Tier 2 treatments? (Updated 3-24-20)
A: Only treatments initiated AFTER an abnormality has begun which begins an episode or Scenario should be entered on the case report form (CRF) as treatments included in the study protocol. (See Exception below). The CRF will document the time of the new episode, and any new treatment initiated. If the treatment was already being actively used before the episode started, it should NOT be entered on the CRF as a new treatment. Treatments already being actively used when an episode occurs should be noted in the medical record, but NOT listed as a new treatment initiated in response to the abnormality for the study. Select another treatment (not already in place) from Tier 1 to address the new episode or select from Tier 2 if Tier 1 treatments are already actively being used and the treatment team chooses not to do another treatment from Tier 1. Note this information and rationale.
Examples:
- If the head of bed is already elevated when the ICP increases to begin a new episode, the raising head of bed is not a new treatment unless the head of the bed is raised further.
- If any intervention is initiated for an episode of elevated ICP and the ICP responded, then that episode has resolved in response to the treatment(s) given. This is documented in the CRF. If a few hours later, there is another episode of elevated ICP, the team can choose to give any Tier 1 treatment again, including the agent given for the earlier episode, if it is thought to be clinically indicated. This is considered a new intervention for a new Scenario or episode.
- If you are doing a treatment that says “ensure temperature” or “maintain a temp”, you must note WHICH treatment was INITIATED in order to “ensure” or “maintain
Exception: If the treatment team is already working in Tier 2 when a new Episode begins, and the clinicians deem it unreasonable or inadvisable to re-visit Tier 1 treatments (e.g., HOB is already elevated, CSF drainage utilized, and others), then it is acceptable NOT to begin treatment of the new Episode with Tier 1 choices.
WebDCU will allow this by doing the following:
Leave Tier 1 interventions empty and click 'Move to Tier 2' for the response to 'Tier 1 results'. This will trigger missing data warnings that can be dismissed by clicking the pencil icon and explaining that Tier 1 treatments are already on board. This will allow the Tier 2 interventions to be entered.
Q: How will sites be rolling out and how many sites will participate in the study?
A: While taking all sites into consideration, we will be prioritizing sites based on experience with placement and management of PbtO2 monitors. We will be contacting the highest priority sites no later than September 10. BOOST will only include sites that use PbtO2 monitors. For sites who do not currently have or use PbtO2 monitors but intend to, you will need to demonstrate experience in using brain tissue oxygen monitors for patient care in TBI patients. Our expectation is that sites should have successfully placed and used PbtO2 monitors at least 3 times in the past 6 months to be considered as a BOOST site. After your site has placed at least 3 PbtO2 monitors, the site PI should contact Dr. Shutter (shutterla@upmc.edu) to confirm this information. We encourage all sites to begin preparations now, and will move forward with sites as they complete this process and are ready to get started. We budgeted for 45 actively enrolling sites in BOOST3.
Q: Does my site have to use the Central/Single IRB?
A: Yes. BOOST-3 is an NIH-funded study awarded after Jan. 25, 2018. The NIH requires mandatory use of a single IRB for all SIREN trials (NIH Policy).
Q: What is the Central IRB #?
A: Pro00030585
Q: What will be involved in the process of ceding to the central IRB (Advarra)?
A: Consistent with NIH guidelines, BOOST 3 will be using a central IRB. The answer to this question and others about cIRB can be found on the following SIREN page: Resources and Steps for Relying on the SIREN ER-CIRB
Q: My site IRB uses Smart IRB for reliance agreements. How does that work?
A: Please contact your IRB for assistance with this. You can also reference instructions from Advarra: Using Smart IRB
Q: What is expected of each site for the EFIC process for this trial?
A: Sites with experience conducting EFIC activities can expect to propose community consultation and public disclosure (CC/PD) activities similar to the efforts undertaken in previous trials. However, the cIRB may yet request the overall trial EFIC plan to have some additional or different requirements. An EFIC plan for sites to use to build out your local CC/PD plan will be made available as soon as it has been cIRB approved. Centrally, the CCC will create CC/PD related resources and templates for local use, e.g, videos, brochures, ads, flyers, slide sets, self-administered survey instruments, etc. CC and PD results and event data will be collected centrally in WebDCU™ for generating reports for the cIRB. There will be startup funds allocated to perform the required EFIC activities. If you have additional questions please contact Deneil Harney dkolk@umich.edu 734-232-2132
Q: At what point is it permissible to enroll a patient under EFIC?
A. An eligible patient can immediately be enrolled at the time of probe placement if the LAR is not available. According to the Protocol (Section 4.3.2 Recruitment and informed consent): “Upon hospital arrival of a potentially eligible subject, study teams will diligently try to determine the availability of an LAR. If an LAR is available prior to the successful placement of intracranial monitoring, the patient will only be enrolled with the written informed consent of the LAR. If an LAR is not available prior to the successful placement of intracranial monitoring, eligible patients will be enrolled with EFIC immediately after intracranial monitor placement, as BTF and ACS guidelines recommend starting intracranial monitoring as promptly as practically possible. Subsequent to an EFIC enrollment, efforts to contact an LAR will continue. An LAR will be notified of an EFIC enrollment and consent to continue in the study will be sought at the earliest opportunity.”
Attempts should be made to identify and contact the participant’s LAR and should be documented in the Informed Consent Log in the BOOST-3 Database/WebDCU.
Q: If a family member is not available to provide consent, but is available on the phone can the patient be enrolled under EFIC?
A:You must discuss the study with the LAR on the phone if the LAR is available on the phone. If they say yes and can’t be physically available and cannot consent via e-consent prior to probe placement then you can enroll under EFIC. If the LAR refuses enrollment in the study the patient should not be enrolled.
Q: How does the possibility of getting a remote e-consent affect the rule about who can be enrolled with EFIC?
A: The potential to get a remote e-consent does not affect the BOOST Protocol EFIC rule about who can be enrolled with EFIC (No family present prior to probe placement). As always, if there is no LAR available on site to consent for enrollment, there should still always be an attempt to locate an LAR or other family member by telephone. If an LAR or family member can be reached by telephone, the study team needs to find out if they object to participation. If the LAR or family member objects, the patient cannot be enrolled with EFIC. If they do not object and all other necessary conditions are met, the patient may be enrolled with EFIC.
If there is the ability and time available to do remote e-consent instead of an EFIC enrollment, and that is what the LAR wants to do, then e-consent can be used for prospective informed consent.
For LAR and family members that are never expected to be available in person, the remote e-consent can also be used after an EFIC enrollment to consent for continued participation.
Please consult the CRF Completion guidelines, pg. 19 Form 211, for detailed instructions on documenting your ongoing attempts to locate family, obtain no objection and subsequently their consent decision on the ICL-CRF.
Q: The timestamp on the e-consent does not reflect my time zone. What should I do? (Updated 3-3-20)
A: A generic note to file is now available that reflects clarification on the time zone reflected on the e-consent PDF and the solution moving forwarded.
Q: How will BOOST 3 data be managed?
A: The BOOST 3 database is currently being built in the WebDCU™ clinical trial management system. WebDCU is housed at the Data Coordination Center (DCC) at the Medical University of South Carolina (MUSC). All trial data will be maintained in this secured system, including CRFs, regulatory requirements, and EFIC CC and PD activities. The central IRB application process will also take place in this system. Training and instructional materials will be provided to facilitate use of each component of the database.
Q: Which CRFs need to be completed and entered into WebDCUTM prior to randomization?
A: The user must first complete and submit in order: the Subject Enrollment, baseline Glasgow Coma Scale (Form 138), and the Inclusion and Exclusion Criteria (Form 101) forms, with all eligibility criteria met. Once these forms have been submitted, the user will then complete and submit the Randomization form (Form 102).
Q: How should screened patient be entered into the WebDCU Screening Log during the COVID19 enrollment hold? (New 3-25-20)
A: Sites should continue to screen as they normally would and indicate the reason(s) why the subject was not eligible. If the subject is otherwise eligible for the trial, then the site will select “other reason” and enter “COVID19 hold”.
Q: How will local site training take place?
A: Sites will be responsible for training their local study teams and clinical staff initially and throughout the course of the trial. The SIREN CCC will provide training materials, available online for easy access (protocol training, in-service slides, etc.), as well as study materials to be distributed locally (e.g. pocket cards, trial posters, etc.). As sites develop their own materials, we encourage sharing across the network and can post these materials in the BOOST 3 Toolbox.
Equipment (Moberg, Pbt02, etc.)/Trial Materials
Q: The Moberg CNS isn't detecting my USB drive, or I can't see the archived data when connecting it to my computer. (New 11/17/23)
A: The CNS Monitor does not support software-encrypted USB drives. An increasing number of hospitals automatically load software encryption on USB drives, rendering them undetectable and unusable by the CNS Monitor. If the CNS Monitor says that it can't detect a USB drive, it may be encrypted.
Our recommendation is to maintain a separate dedicated USB drive that is only connected to the CNS Monitor and a computer capable of reading the unencrypted USB drive for data offload. Sometimes that may mean a personal/non-institutional computer. That USB drive should be formatted as FAT/FAT32 or NTFS and must not use any software encryption.
If your hospital IT policy prohibits using unencrypted USB drives, the following hardware-encrypted USB drives (unlocked by entering a pin on a physical keypad on the USB drive itself) have been tested and confirmed to work with the CNS Monitor:
- Aegis Padlock DT 3.0
- Aegis Apricorn Padlock drive
- Aegis Apricorn Secure Key
Even if you use one of those drives, you should still not connect it to a hospital computer that does not allow reading from unencrypted drives, as it may still be detected as an unencrypted drive after entering the pin. A hospital computer that does not allow reading from unencrypted drives may ask to, or may automatically, try to encrypt the drive. Loading encryption software on any USB drive will make the drive unusable on the CNS Monitor.
Q: What trial equipment will be provided to sites?
A: The SIREN CCC will be lending Moberg CNS monitors (CNS-310) to all enrolling sites. So long as sites continue to enroll, the devices will remain on site. Should sites discontinue enrollment, the devices will be returned to the SIREN CCC. Pbt02 monitors will NOT be provided by the SIREN CCC. For enrolling sites, startup payments will include some funds to partially offset the cost of site’s PbtO2 ownership, and may be applied to equipment necessary to connect the PbtO2 device (Integra Licox or Raumedic Neurovent) to the Moberg monitor. The cost of probes is built into per subject payments.
Q: What do you do if it appears that the Pbt02 monitor is not functioning properly?
A: If there is evidence that a probe is not working and a first and second Fi02 challenge have failed, follow the next steps as outlined in the MOP (CT and replacement of probe if indicated but only in the unblinded group). In the blinded group, there is no remedy. However, you muct do and FiO2 challenge daily unthe 5th day after placement or the probes are removed.
Open Pbt02 treatment are: If, after replacement of the probe, the new probe still fails the FiO2 challenge, the team may then choose to ignore the PbtO2 numbers. The protocol does not require replacing the probe a second time. If Fi02 challenges in subsequent days show that the probe starts working (i.e., the FiO2 challenge is successful), interventions for low PbtO2 can be re-started.
In all cased the primary analysis will be performed using intention to treat principles.
Q: What is the best way to silence alarms on the Moberg at the start of an episode?
A: When an episode starts, the alarm will need to be acknowledged through the CNS Reader. If currently viewing the CNS Monitor desktop, a pop-up message will appear on screen prompting the user to ”Switch Desktop”.
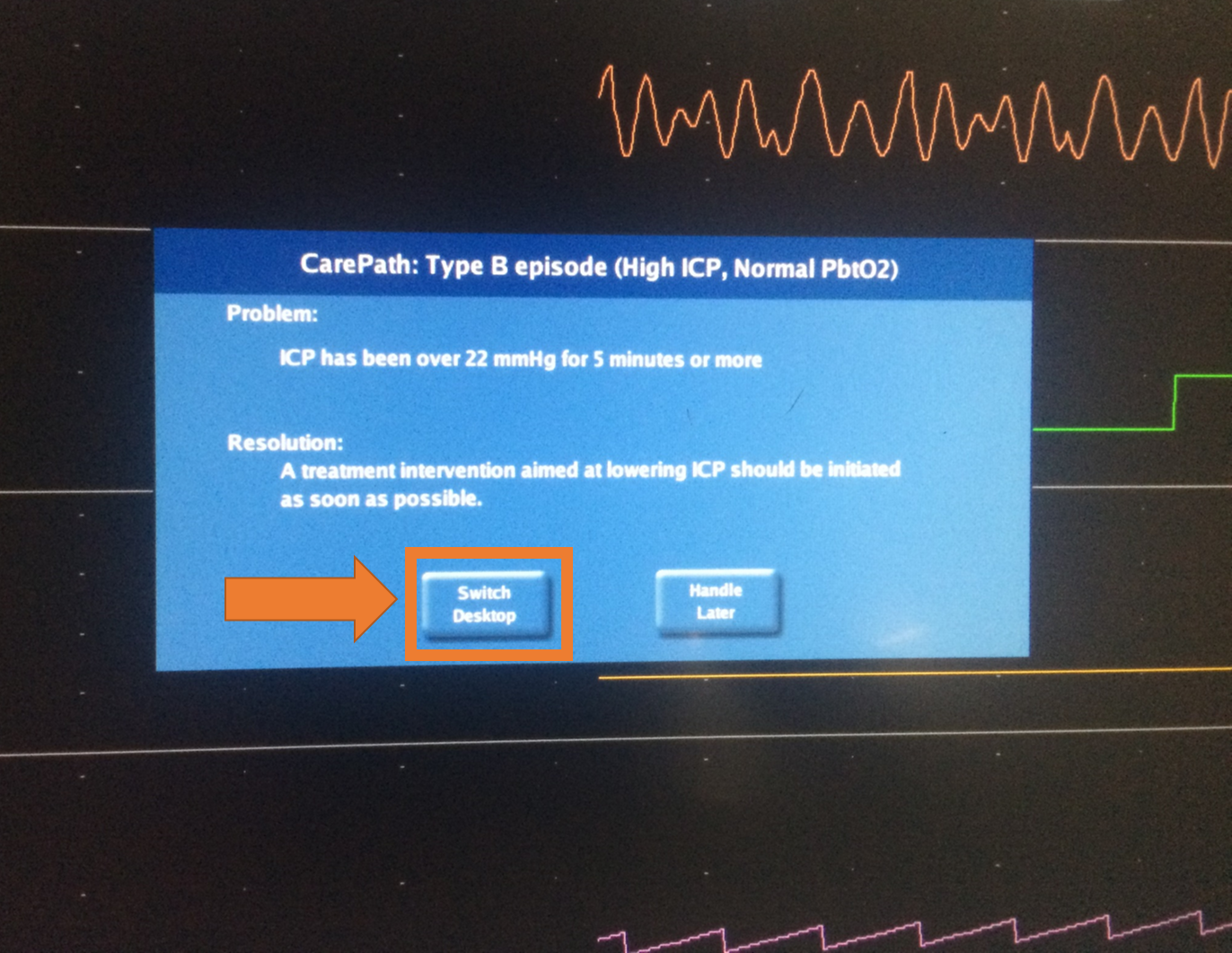
Press ”Switch Desktop” in order to switch to CNS Reader/CarePath. When viewing the CNS Reader/CarePath at the beginning of an episode, a pop-up message will appear on screen showing the “INTERVENTION REQUIRED Press to Acknowledge” button.

Q: An alarm went off but it is not associated with an episode. How to silence it?
A: If data collection is ongoing but CarePath is not running and analyzing the data to identify episode, an alarm will go off. If currently viewing the CNS Monitor desktop, a pop-up message will appear on screen prompting the user to ”Switch Desktop”.

Press ”Switch Desktop” in order to switch to the Windows desktop. On the Windows Desktop:
- Ensure that the CNS Reader application is running. If not, double-click on the CNS Reader Admitted shortcut to launch it.
- Within the CNS Reader application, go to the ‘Layouts’ menu on the menu bar and confirm that the currently selected layout is called ‘CarePath’ (as denoted by a checkmark on its side).
- Click on the Summary tab on the left-hand side to select the summary page. Confirm that Display 1: Summary is visible; if not, click on the “Display 1” button on the application toolbar. Check whether Display 1: Summary shows “PROTOCOL NOT RUNNING”.

In that case, click on the CarePath tab on the left-hand side to switch to the flowchart view. From the “Setup Monitors” heading, confirm that both ICP and PbtO2 monitors are in place by clicking the “ICP Placed” and “PbtO2 Placed” buttons.

Q: Can the brain oxygen monitor be connected to the vital signs monitor?
A: Not for data collection during the BOOST-3 study. Direct connection to the CNS Monitor is needed for two main reasons:
- It is the only way to ensure data blinding at the bedside (the vital signs monitor does not blind PbtO2) and to ensure that no brain oxygen data is stored in the EMR, available to everyone.
- It is the only way the alarming software (CarePath) will recognize the brain oxygen data.
Q: What is the best way to silence alarms on the Moberg at the start of an episode when running older CNS Reader/CarePath software (prior to Z.00.28)? NOT RECOMMENDED
A: Ideally, the Moberg display should always be on the Summary Screen in the CNS Reader, as shown in the following screenshot:


In the event of a technical Moberg issue, please contact Moberg support at: support@moberg.com. If this an emergent technical issue, please call the BOOST-3 Emergency Hotline at: 855-4-BOOST3 (855-426-6783).
Q: How is Moberg data uploaded at the end of participant monitoring?
A: Upon discharging the patient, the CNS Monitor offers options for managing the collected patient data. To create a permanent record of patient data on external media, press Archive Now. To keep the session on the monitor until you are ready to archive, press Archive Later.
- Archive Now: Pressing Archive Now gives the user the option to archive collected data to an external USB drive. The user will be instructed to insert the USB drive. Once the USB drive has been verified, archiving will begin and a moving status bar will indicate the progress of archiving. A dialog box will appear once archiving is complete.
- Archive Later: Pressing Archive Later saves the patient file on the CNS Monitor for review or archiving at a later time. Saved files can be found by pressing Manage Data Files on the CNS opening/start-up screen. Selecting the desired patient provides information about the file and allows the user to Review, Archive, or Erase data.
To Upload Data to the IBM Cloud: A video on how to move the Moberg data into the IBM cloud account is posted in the BOOST-3 website’s Toolbox under the Moberg Device link.
Q: What if my patient is off unit and we are not able to capture continuous data on the Moberg monitor?
A: Based on the BOOST3 MOP, “Periods when such data is artifactual as a consequence of the patient being disconnected from the monitors for transport to an imaging study or other procedure should be annotated using software tools in the Moberg CNS Monitor by the research team…” (page 51). If the patient is undergoing a surgical procedure, data collection will be temporarily interrupted during that time until the patient returns to the ICU; an annotation should be made in CarePath indicating the interruption of data collection. Annotations to explain interruptions should be selected from the available list under the ‘Monitor’ heading. Research coordinators should review the clinical chart and query bedside nurses to confirm times when the patient was disconnected and identify explanations for periods of artifactual data.

Q: How do I know if the Moberg CNS Monitor has the latest BOOST3 software update?
A: Below are instructions for sites to check that they have the latest version of software installed:
On the CNS Monitor
- From the opening screen on the CNS Monitor, press System Configuration-->System Information.
- Confirm that the software version is B.15.27
On the Application PC (CNS Reader/CarePath)
- From the opening screen on the CNS Monitor, press System Configuration --> System Information --> Application PC --> View Desktop. This will switch the view to the Windows operating system running on the Application PC.
- Double click on the launch icon called 'CNS Reader' (NOT the 'CNS Reader Admitted') to run the CNS Reader application.
- In the Help menu, select About CNS Reader.
- Confirm that the software version is Z.00.29.
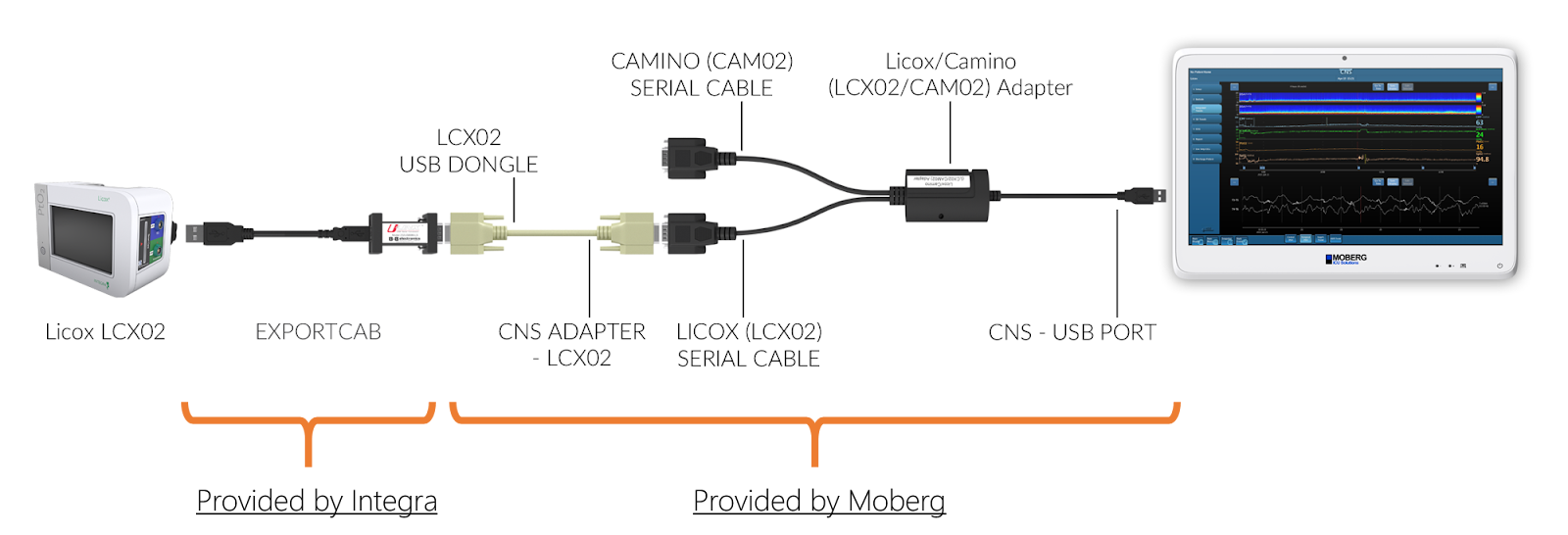
Q: How do I connect the new Licox (LCX02) to the Moberg CNS Monitor?
A: The new Licox models (LCX02) require cables provided by Moberg AS WELL AS the USB-to-serial adapter provided by Integra, called EXPORTCAB. Without the EXPORTCAB no data can be collected from the Licox LCX02.
Connect the EXPORTCAB to the USB port on the back of the Licox LCX02. Then connect to it the end labeled “LCX02 USB DONGLE” of the serial cable. The other end, labeled “CNS ADAPTER-LCX02”, needs to be connected to the Licox/Camino Adapter (a Y cable), specifically to the end labeled “LICOX (LCX02) SERIAL CABLE”. Then connect the USB end of the Y cable to any of the USB ports on the bottom of the rear panel of the CNS Monitor.
Q: Can I collect more than ICP, PbtO2, blood pressure, and temperature?
A: Yes. While ICP, PbtO2, blood pressure, and temperature are the main study-related variables, you can (and are encouraged to) collect additional variables.
When no patient is admitted on the CNS Monitor (i.e. there is no ongoing monitoring session), select “System Configuration” from the opening screen, then press “Select Devices”. Confirm that all devices that will be connected to the Moberg CNS Monitor are selected (if they are selected, the selection box will be green). If they are not selected, press on their respective selection boxes on their left.
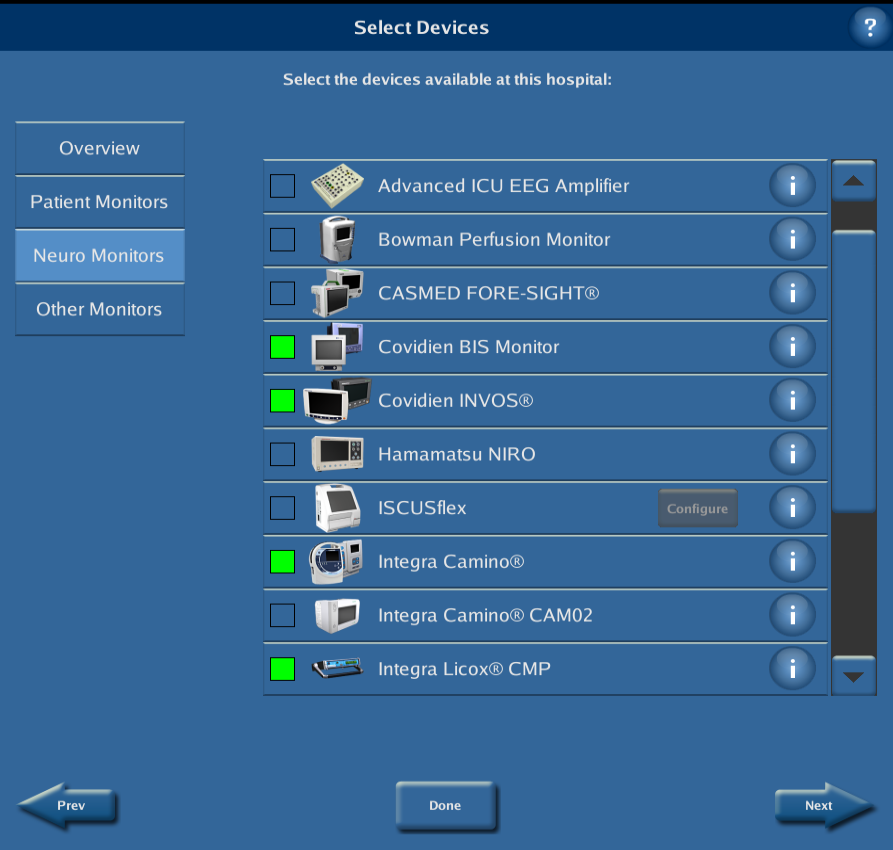
Press “Done” to close the Select Device dialog. After the device selection process is complete, the data collection protocol needs to be customised to include the measurements of interest. To this end, press “Protocols” from the System Configuration Menu. Select the BOOST-3 protocol to be edited and press “Edit Protocol”. (If prompted, Select Study Measurements, then press “Accept”.) Press on “Select Measurements” and identify a source for all the measurements that you would like to collect from the external devices, ensuring that you select the same measurement labels as the ones that are displayed on the external devices (e.g. ICP vs. IC1, etc.). Note that measurements are organized by category; use the buttons on the left to move between categories. When all selections are complete, press “Done”.
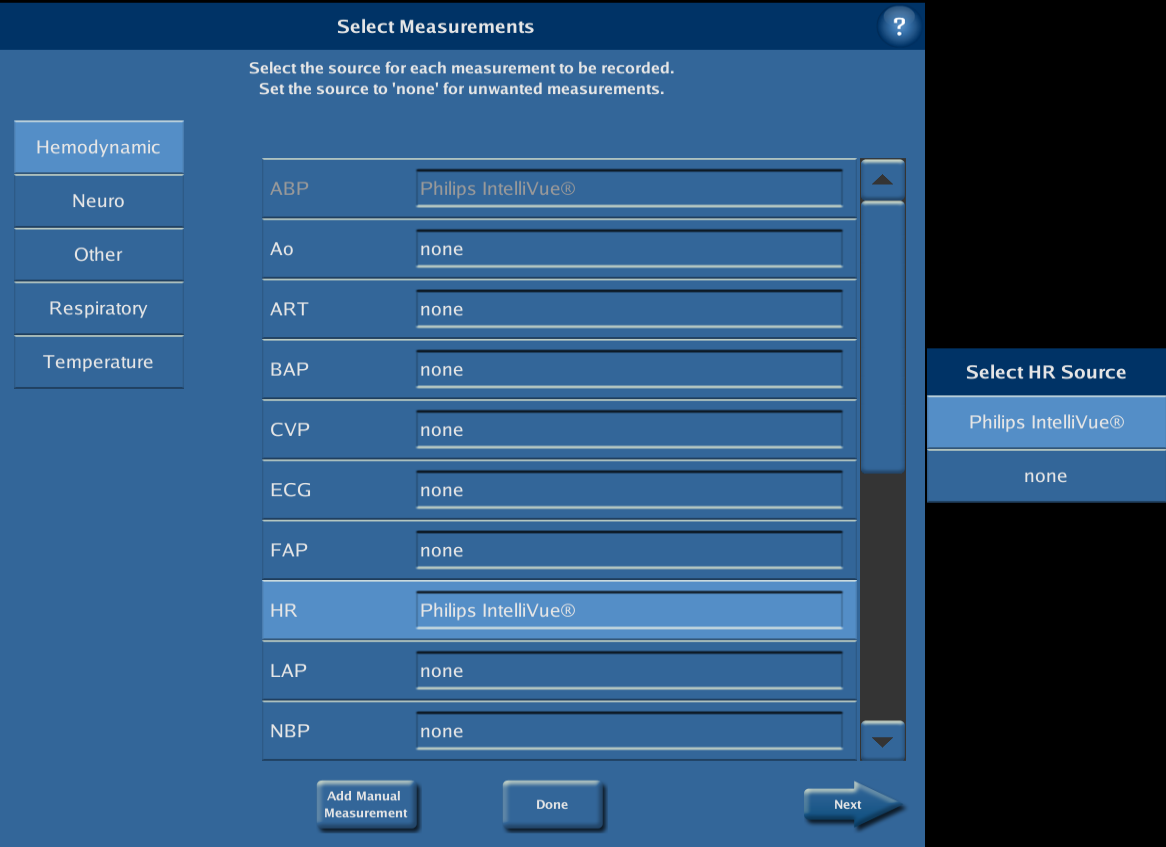
All of the measurements for which a source was identified will now be collected and saved. In order to also show such measurements at the bedside, refer to the technical bulletin “Editing Protocols on the CNS Monitor”. When all changes are complete, press ”OK” and select either “Save” or “Save As”.
Q: How can I switch the ICP alarm source in Moberg/CarePath?
A: There may be cases in which a there may be a need to switch ICP sources for continuous measurement during a monitoring period.
Example—a subject has an open ventriculostomy drain for CSF drainage, plus an intraparenchymal ICP that provides continuous data; unexpectedly, the intraparenchymal ICP monitor suddenly fails during monitoring; the ventriculostomy can be closed to allow for continuous measurement, but the CNS Monitor/CarePath alarms will need to be assigned to the closed ventriculostomy ICP source.
When you need to switch to a new ICP measurement to drive CarePath, you can discharge the patient on the CNS Monitor (i.e. end the monitoring session) and then re-admit the patient (i.e. start a new monitoring session). When readmitting, you would then select the new ICP measurement as part of the initial BOOST-related dialog. After starting the new monitoring session, you would also need to re-start Carepath by clicking on CNS Reader Admitted and following the usual steps.
*refer to the ‘Start monitoring’ video in the Toolbox on the BOOST-3 SIREN webpage for detailed steps*
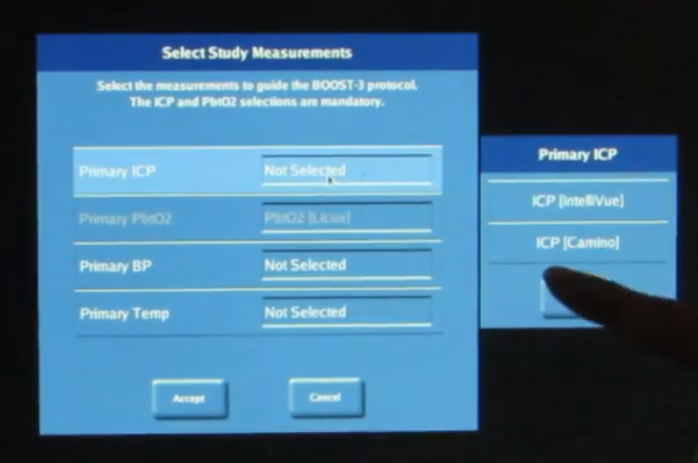
Q: How can I prevent potential unblinding when intracranial monitors are reconnected to the Moberg CNS Monitor (after a patient comes back from a procedure)?
A: Note that this is a concern in the ICP ONLY treatment arm, and for sites that utilize the new Licox monitor (LCXO2):

configurations:
- A site has the LCXO2 and the new Camino --> the recommendation is for the site to connect the new Camino to the vital signs monitor, therefore removing the source of possible confusion. (This may also allow for ICP input directly to their EMR, depending on the site’s EMR/vital signs monitor.) At that point the notification can be safely turned off (see instructions below).
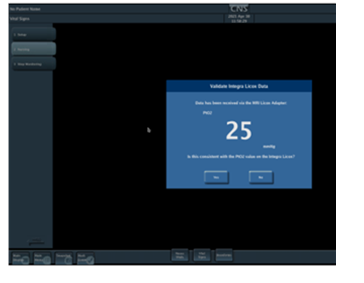
- Alternatively, if the site chooses to connect both the Licox and Camino directly to the CNS monitor—the study coordinator will have to ensure that values match with a visual check either daily or anytime after a patient is disconnected from the CNS monitor).
- A site has the LCXO2 and the old Camino: -->

There is no possible confusion between the two devices, therefore the notification can be safely turned off (the site can still choose to connect the old Camino to the vital signs monitor instead).
- If have a Raumedic, or old Licox, there is no need to do anything.
The LCXO2 should always be connected directly to the CNS (just like the other sources of PbtO2).
Instructions to disable the confirmation dialog box :
- When no patient is being monitoring, press System Configurations in the opening screen.
- Press System Admin and enter the Admin password (default: moberg) and press OK.
- Press Configure Preferences
- Press Integra Data Validation and select Disabled
- Press Done and then Exit Menus to return to the opening screen.
Please note that if this is done—the study coordinator MUST perform a daily visual check on BOTH the CNS Monitor and an uncovered Licox monitor. This can be done by TEMPORARILY unblinding the CNS monitor to visualize PbtO2, while uncovering the Licox to ensure that the values match. This should be done, at minimum, when a subject is disconnected from the CNS monitor and intracranial monitors have been reconnected.
Q: What if the Moberg CNS system alarms are not working?
A: If the monitoring session was not opened using the "CNS Reader Admitted" shortcut (rather, just "CNS Reader") then the monitoring session was not monitored by the software for abnormalities and would not have alarmed. If the "CNS Reader Admitted" shortcut was used, the title bar of the software will show the patient info as entered on the CNS Monitor and the admission time. If not, the CNS Reader should be closed, then the "CNS Reader Admitted" shortcut should be opened. Always use the "CNS Reader Admitted" shortcut.
Q: What does this File Error message on the Moberg CNS screen indicate and what should I do?

A: If this error is seen at any time, it means that
- the Events.xml file is locked,
- the software can’t write to it, and
- the events will appear in the software but will not be saved.
The only way to resolve this issue is by
- shutting down the CNS Monitor entirely,
- unplugging it from the wall after the power LED goes out,
- plugging it back in,
- turning it back on,
- and readmit the patient as usual.
Don't leave the BOOST system running, but rather shut it down after the patient is discharged, then power it up when ready to enroll a new patient.
Q: How can you do an FiO2 challenge if the ventilator is already set at 100% FiO2?
A: Consider performing a blood gas to determine the safety of lowering the FiO2 temporarily for the FiO2 challenge.
- If lowering FiO2 below 100% results in lowering PbtO2 by >5 mmHg, raise FiO2 to 100% again. If that results in increase in PbtO2 by at least 5 mmHg again, that is considered passing the FiO2 challenge.
- If it is not feasible to lower the FiO2, consider doing a MAP or CO2 challenge to determine if that raises the PbtO2. A MAP or CO2 challenge should not be performed in subjects in the ICP-only group.
- If the second FiO2 challenge again fails, further management will be determined by patient group.
- In the ICP+PbtO2 (intervention) group, a non-functioning or mal-positioned PbtO2 probe should be replaced within 2 hours if at all possible.
- In the ICP only (control) group, the study team will document that the PbtO2 probe is unreliable but will not replace it. Do not inform the treatment team that the probe is malfunctioning in a blinded patient.
- You may check the FiO2 daily in case it begins to function. This happens.
Subject Reimbursement/Finances
Q: What is the per subject reimbursement?
A: The per subject reimbursement has not yet been finalized. However, it is expected to be approximately $12,500, which is inclusive of indirects. Per subject reimbursement will be milestone driven.
Q: Are we billing insurance for the procedure?
A: Yes. Placement of probes are considered routine care of TBI patients at all study sites.
Q: Are we billing insurance for the device?
A: The ICP probe is standard of care and the cost of the probe should be billed to the 3rd party payors. The PbtO2 probe is the target of the study, and the cost of the probe is included in the per-patient capitated cost. There is no charge for the use of the Moberg CNS monitor.
Q: Will the sponsor pay for the device or procedure if insurance denies?
A: No. As placement of the probes is part of routine care, the cost for the procedure to place the probes and the ICP probe itself should be billed to insurance. The cost of the PbtO2 probe is covered by the sponsor. There is no cost associated with the monitoring device. As a reminder: Please discuss your local practices for intracranial monitoring with the treating neurointensivist or neurosurgeon at your site.
Q. What is the management strategy for the BOOST-3 trial?
A. BOOST-3 is a trial assessing a management strategy of patient care; it is NOT a study assessing an experimental drug, device or procedure. The procedures being done for this trial, including placement of intracranial monitoring probes, is part of the routine care for patients admitted to the hospital (i.e. inpatients) with severe TBI.
Q. Do you have the IDE #?
A. No, there is an IDE waiver from the FDA since PbtO2 devices are FDA-cleared for this indication.
Q. Where can I get more information on Integra/Raumedic products or their IFUs?
A. Integra: http://www.integralife.eu/products/neuro/neurocritical-care/pbto2-licox/
Raumedic: https://www.raumedic.com/us/neuromonitoring/neuro-icu/oxygen-partial-pressure/